LOTIS-2 was a large registrational trial in r/r DLBCL therapy (N=145)1
Open-label, single-arm phase 2 study design1,2

Key DLBCL inclusion criteria1,2
- ASCT-eligible and -ineligible
- DLBCL NOS
- DLBCL arising from low-grade lymphoma
- HGBCL with MYC and BCL2 and/or BCL6 rearrangements
- ECOG 0-2
Key DLBCL exclusion criteria1
- Bulky disease (≥10 cm)
- Lymphoma with active CNS involvement
aIn LOTIS-2, patients received 0.15 mg/kg for the first 2 cycles, then 0.075 mg/kg for subsequent cycles.1 The average weight was 77.1 kg, requiring 2 vials for the first 2 cycles, then 1 vial for subsequent cycles.3
bPET-CT evaluated by independent review committee using Lugano 2014 criteria.1
ASCT = autologous stem cell transplant; CNS = central nervous system; CR = complete response; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; ECOG = Eastern Cooperative Oncology Group; HGBCL = high-grade B-cell lymphoma; NOS = not otherwise specified; ORR = overall response rate; PET-CT = positron emission tomography-computed tomography; PR = partial response; r/r = relapsed/refractory.
LOTIS-2 included a broad range of heavily pretreated patients with difficult-to-treat disease1
Select baseline patient and disease characteristics (N=145)1,a
|
|
|
|
|
|
|
|
|
|
Broad study population reflects the disease’s heterogeneity
aData from 2-year follow-up; may differ from those in primary analysis.4
bAlternatively referred to in LOTIS-2 as transformed DLBCL; the diagnosis of DLBCL NOS included DLBCL arising from low-grade lymphoma.1
cHGBCL with MYC and BCL2 and/or BCL6 rearrangements.2
dABC and GCB were investigator-reported without independent testing; unknown 51%.2
ePrimary refractory defined as no response to front-line therapy.3
fAs defined by ADC Therapeutics, intensive salvage therapy includes one or more of the following: chemo-immunotherapy (ICE, GDP, DHAP, DHAX; all generally combined with rituximab), SCT, and/or CAR-T.3
ABC = activated B-cell; CAR-T = chimeric antigen receptor T cell; DHAP = dexamethasone, cytarabine, cisplatin; DHAX = dexamethasone, cytarabine, oxaliplatin; GCB = germinal center B-cell; GDP = gemcitabine, dexamethasone, cisplatin; ICE = ifosfamide, carboplatin, etoposide; SCT = stem cell transplant.
Meaningful response with ZYNLONTA® as a single agent1
Nearly half (48.3%) of patients achieved a response
Primary analysis
(N=145)1,a
ORR 48.3%
(primary endpoint)
(95% CI: 39.9, 56.7)
CR 24.1%
(n=35)
(95% CI: 17.4, 31.9)
PR 24.1%
(n=35)
(95% CI: 17.4, 31.9)
aMedian follow-up: 7.3 months (range: 0.3-20.2).1
2-year
follow-up4,b
bPatients were followed
for 2 years after study
completion. Median
follow-up, all patients:
7.8 mos (range: 0.3-42.6).4
cMedian follow-up, CR
patients: 35 mos
(range: 4.4-42.6).5

CI = confidence interval; CR = complete response; ORR = overall response rate; PR = partial response.
Median time to response with ZYNLONTA® as a single agent1,3,5
Primary analysis
(N=145)1
OR 1.3 Months1,a
(n=70)
(range: 1.1-8.1)
CR 1.4 Months3,b
(n=35)
(range: 1.2-14.0)
During the treatment period, imaging was performed at 6 weeks and 12 weeks after Cycle 1, Day 1, then every 9 weeks until end of treatment.2
2-year
follow-up3,4,c
cMedian follow-up, all patients:
7.8 mos (range: 0.3-42.6).4
dMedian follow-up,
CR patients: 35 mos
(range: 4.4-42.6).5
Median time to OR (n=70)

1.3 Months
(range: 1.1-8.1)
Median time to CRd (n=36)
1.5 Months
(range: 1.2-14.0)
aMedian follow-up time: 7.3 mos (range: 0.3–20.2).1
bExploratory data: LOTIS-2 was not designed or powered to evaluate rapidity of CRs. Consider small sample size when interpreting results.
OR = overall response.
Durable responses in 2-year follow-up1,4,b-e
Primary analysis
(N=145)1,a
mDOR of OR (n=70)1
10.3 Months
(95% CI: 6.9, NE)
mDOR of CR (n=35)2,a,b
13.4 Months
(95% CI: 10.3, NE)
mDOR of PR (n=35)2,b
5.7 Months
(95% CI: 1.7, NE)
aMedian follow-up: 7.3 months (range 0.3–20.2). Of 70 patients with an OR, 25 (36%) were censored prior to 3 months; 26% of responders had a DOR of ≥6 months.1
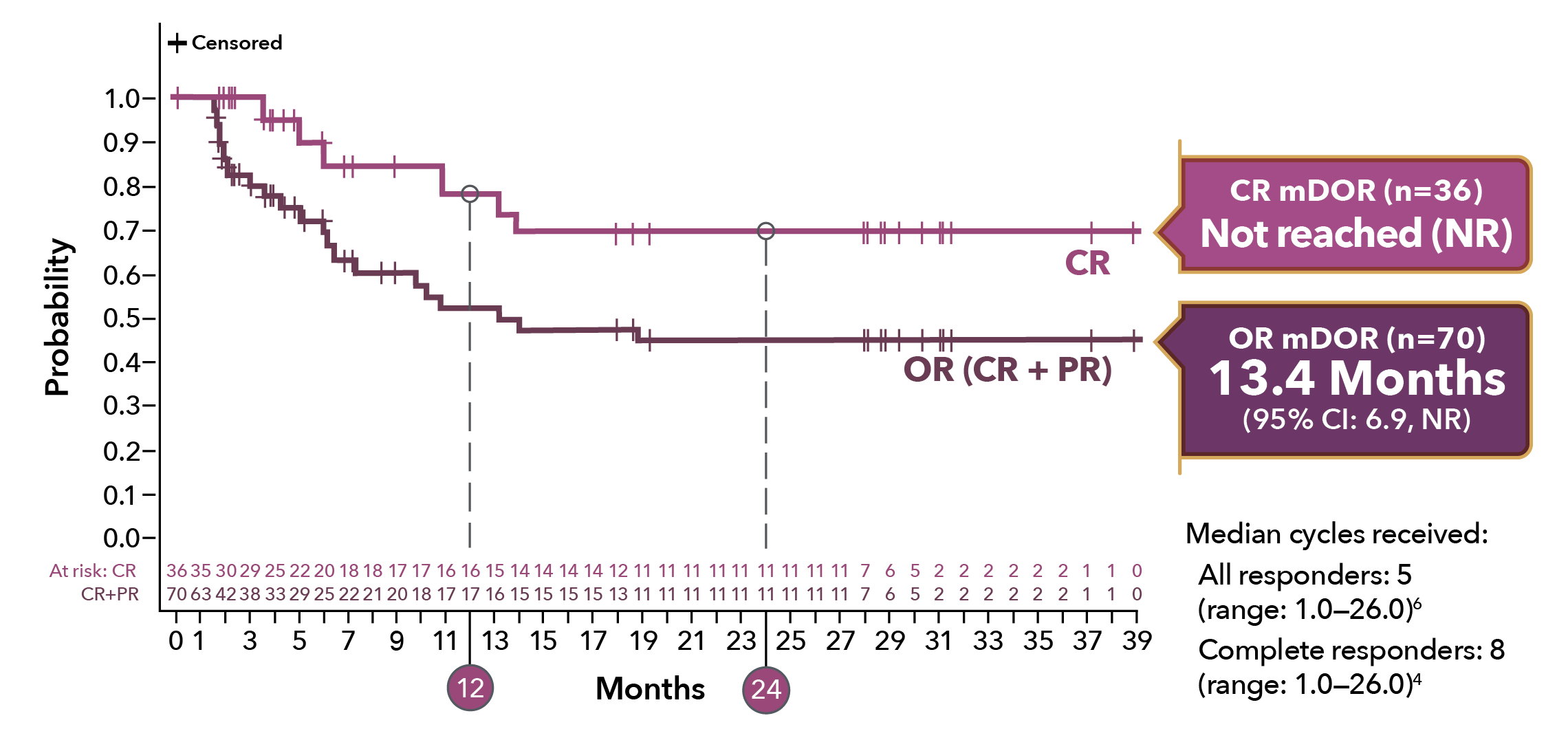
dMedian follow-up, all patients: 7.8 mos (range: 0.3-42.6).4
eMedian follow-up, CR patients: 35 mos (range: 4.4-42.6).5
bExploratory data: LOTIS-2 was not designed or powered to evaluate duration of CRs or PRs. Consider small sample size when interpreting results.
cOf 36 patients with a CR, 7 (19%) were censored prior to 3 mos; 56% of patients with a CR had a DOR of ≥6 mos.4
mDOR = median duration of response; NE = not estimable.
Response in select patient subgroups3,4
Exploratory analyses of primary (ORR) and secondary (CR) endpoints in predefined subgroups.3
Subgroup analyses were not designed or powered to demonstrate an effect in patient subgroups. Consider small sample size when interpreting results.3
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
68 patients received subsequent anti-cancer therapy, including2,4
- 12 patients who went on to consolidation with SCT
- 16 patients who received CAR-T therapy after disease progression
- In a retrospective subanalysis of LOTIS-2 patients who went on to CAR-T (n=14), 10 of 10 patients who were tested all had CD19 antigen expression after ZYNLONTA® treatment.
The safety and efficacy outcomes for these patients have not been confirmed with a randomized clinical trial.2
an = responders.

References: 1. ZYNLONTA [package insert]. Murray Hill, New Jersey: ADC Therapeutics SA; 2026. 2. Caimi PF, Ai W, Alderuccio JP, et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS 2): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021;22(6):790-800. 3. Data on file. ADC Therapeutics SA. 4. Caimi PF, Ai WZ, Alderuccio JP, et al. Loncastuximab tesirine in relapsed/refractory diffuse large B-cell lymphoma: long-term efficacy and safety from the phase II LOTIS-2 study. Haematologica. 2024;109(4):1184-1193. 5. Caimi PF, Ai WZ, Alderuccio JP, et al. Long-term responses with loncastuximab tesirine: updated results from LOTIS-2, the pivotal phase 2 study of patients with relapsed/refractory diffuse large B-cell lymphoma. Poster presented at: European Hematology Association 2023 Hybrid Congress. Frankfurt, Germany and virtual, June 8-11, 2023. 6. Zinzani PL, Caimi, PF, Carlo-Stella C, et al. LOTIS-2 follow-up analysis: updated results from a phase 2 study of loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma. Poster presented at: 16th International Conference on Malignant Lymphoma, Virtual Edition, June 18-22, 2021.