HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ZYNLONTA safely and effectively.
See full prescribing information for ZYNLONTA.
ZYNLONTA®(loncastuximab tesirine-lpyl) for injection, for intravenous use
Initial U.S. Approval: 2021
Indications and Usage
ZYNLONTA is a CD19-directed antibody and alkylating agent conjugate indicated for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low-grade lymphoma, and high-grade B-cell lymphoma. (1)
This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s). (1)
Dosage and Administration
- ZYNLONTA is an intravenous infusion over 30 minutes on Day 1 of each cycle (every 3 weeks). The recommended dosage is:
- 0.15 mg/kg every 3 weeks for 2 cycles.
- 0.075 mg/kg every 3 weeks for subsequent cycles. (2.1)
- Premedicate with dexamethasone 4 mg orally or intravenously twice daily for 3 days beginning the day before ZYNLONTA. (2.2)
- See Full Prescribing Information for instructions on preparation and administration. (2.4)
Dosage Forms and Strengths
For injection: 10 mg of loncastuximab tesirine-lpyl as a lyophilized powder in a single-dose vial for reconstitution and further dilution. (3)
Containdications
None (4)
Warnings and Precautions
- Effusion and Edema: Monitor for the development of pleural effusion, pericardial effusion, ascites, peripheral edema, and general edema. Consider diagnostic imaging when symptoms develop or worsen. (5.1)
- Myelosuppression: Monitor blood cell counts. Withhold, reduce, or discontinue ZYNLONTA based on severity. (5.2)
- Infections: Monitor for infection and treat promptly. (5.3)
- Cutaneous Reactions: Monitor patients for new or worsening cutaneous reactions, including photosensitivity reactions. Dermatologic consultation should be considered. (5.4)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.5, 8.1, 8.3)
Adverse Reactions
Most common (≥20%) adverse reactions, including laboratory abnormalities, are thrombocytopenia, increased gamma-glutamyltransferase, neutropenia, anemia, hyperglycemia, transaminase elevation, fatigue, hypoalbuminemia, rash, edema, nausea, and musculoskeletal pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact ADC Therapeutics at 1-855-690-0340 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use in Specific Populations
- Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA‑approved patient labeling.
Revised: 10/2022
1. Indications and Usage
ZYNLONTA is indicated for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low-grade lymphoma, and high-grade B-cell lymphoma.
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
2. Dosage and AdminisTration
2.1 Recommended Dosage
ZYNLONTA as an intravenous infusion administered over 30 minutes on Day 1 of each cycle (every 3 weeks). Administer intravenous infusion as follows:
- 0.15 mg/kg every 3 weeks for 2 cycles.
- 0.075 mg/kg every 3 weeks for subsequent cycles.
2.2 Recommended Premedication
Unless contraindicated, administer dexamethasone 4 mg orally or intravenously twice daily for 3 days beginning the day before administering ZYNLONTA. If dexamethasone administration does not begin the day before ZYNLONTA, dexamethasone should begin at least 2 hours prior to administration of ZYNLONTA.
2.3 Dosage Modifications and Delays
Recommended Dosage Modifications for Adverse Reactions
Adverse Reactions |
Severitya |
Dosage Modification |
||
|---|---|---|---|---|
| Hematologic Adverse Reactions | ||||
| Neutropenia [see Warnings and Precautions (5.2)] | Absolute neutrophil count less than 1 x 109/L | Withhold ZYNLONTA until neutrophil counts returns to 1 x 109/L or higher | ||
| Thrombocytopenia [see Warnings and Precautions (5.2)] | Platelet count less than 50,000/mcL | Withhold ZYNLONTA until platelet count returns to 50,000/mcL or higher | ||
| Nonhematologic Adverse Reactions | ||||
| Edema or Effusion [see Warnings and Precautions (5.1)] | Grade 2a or higher | Withhold ZYNLONTA until the toxicity resolves to Grade 1 or less | ||
| Other Adverse Reactions [see Warnings and Precautions (5.3, 5.4), Adverse Reactions (6.1)] | Grade 3a or higher | Withhold ZYNLONTA until the toxicity resolves to Grade 1 or less | ||
- National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0
Recommendations for Dosage Delays
If dosing is delayed by more than 3 weeks due to toxicity related to ZYNLONTA, reduce subsequent doses by 50%. If toxicity reoccurs following dose reduction consider discontinuation.
Note: If toxicity requires dose reduction following the second dose of 0.15 mg/kg (Cycle 2), the patient should receive the dose of 0.075 mg/kg for Cycle 3.
2.4 Reconstitution and Administration Instructions
Reconstitute and further dilute ZYNLONTA prior to intravenous infusion. Use appropriate aseptic technique.
ZYNLONTA is a hazardous drug. Follow applicable special handling and disposal procedures.1
Dose calculation
Calculate the total dose (mg) required based on the patient’s weight and prescribed dose [see Dosage and Administration (2.1)].
- For patients with a body mass index (BMI) ≥35 kg/m2, calculate the dose based on an adjusted body weight (ABW) as follows:
ABW in kg = 35 kg/m2 × (height in meters)2
- More than one vial may be needed to achieve the calculated dose.
- Convert the calculated dose (mg) to volume using 5 mg/mL, which is the concentration of ZYNLONTA after reconstitution.
Reconstitution of lyophilized ZYNLONTA
- Reconstitute each ZYNLONTA vial using 2.2 mL of Sterile Water for Injection, USP, with the stream directed toward the inside wall of the vial to obtain a final concentration of 5 mg/mL.
- Swirl the vial gently until the powder is completely dissolved. Do not shake. Do not expose to direct sunlight.
- Inspect the reconstituted solution for particulate matter and discoloration. The solution should appear clear to slightly opalescent, colorless to slightly yellow. Do not use if the reconstituted solution is discolored, is cloudy, or contains visible particulates.
- Use reconstituted ZYNLONTA immediately. If not used immediately, store the reconstituted solution in the vial for up to 4 hours refrigerated at 2°C to 8°C (36°F to 46°F) or room temperature 20°C to 25°C (68°F to 77°F). Do not freeze.
- The product does not contain a preservative. Discard unused vial after reconstitution if the recommended storage time is exceeded.
Dilution in infusion bag
- Withdraw the required volume of reconstituted solution from the ZYNLONTA vial using a sterile syringe. Discard any unused portion left in the vial.
- Add the calculated dose volume of ZYNLONTA solution into a 50 mL infusion bag of 5% Dextrose Injection, USP.
- Gently mix the intravenous bag by slowly inverting the bag. Do not shake.
- If not used immediately, store the diluted ZYNLONTA infusion solution refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours or at room temperature 20°C to 25°C (68°F to 77°F) for up to 8 hours. Discard diluted infusion bag if storage time exceeds these limits. Do not freeze.
- No incompatibilities have been observed between ZYNLONTA and intravenous infusion bags with product-contacting materials of polyvinylchloride (PVC), polyolefin (PO), and PAB® (copolymer of ethylene and propylene).
Administration
- Administer by intravenous infusion over 30 minutes using a dedicated infusion line equipped with a sterile, non-pyrogenic, low-protein binding in-line or add-on filter (0.2- or 0.22-micron pore size) and catheter.
- Extravasation of ZYNLONTA has been associated with irritation, swelling, pain, and/or tissue damage, which may be severe [see Adverse Reactions (6.1)]. Monitor the infusion site for possible subcutaneous infiltration during drug administration.
- Do not mix ZYNLONTA with or administer as an infusion with other drugs.
3. Dosage Forms and Strengths
For Injection: 10 mg of loncastuximab tesirine-lpyl as a white to off-white lyophilized powder in a single-dose vial for reconstitution and further dilution.
4. Contraindications
None
5. Warnings and Precautions
5.1 Effusion and Edema
Serious effusion and edema occurred in patients treated with ZYNLONTA. Grade 3 edema occurred in 3% (primarily peripheral edema or ascites) and Grade 3 pleural effusion occurred in 3% and Grade 3 or 4 pericardial effusion occurred in 1% [see Adverse Reactions (6.1)].
Monitor patients for new or worsening edema or effusions. Withhold ZYNLONTA for Grade 2 or greater edema or effusion until the toxicity resolves. Consider diagnostic imaging in patients who develop symptoms of pleural effusion or pericardial effusion, such as new or worsened dyspnea, chest pain, and/or ascites such as swelling in the abdomen and bloating. Institute appropriate medical management for edema or effusions [see Dosage and Administration (2.3)].
5.2 Myelosuppression
Treatment with ZYNLONTA can cause serious or severe myelosuppression, including neutropenia, thrombocytopenia, and anemia. Grade 3 or 4 neutropenia occurred in 32%, thrombocytopenia in 20%, and anemia in 12% of patients. Grade 4 neutropenia occurred in 21% and thrombocytopenia in 7% of patients. Febrile neutropenia occurred in 3% [see Adverse Reactions (6.1)].
Monitor complete blood counts throughout treatment. Cytopenias may require interruption, dose reduction, or discontinuation of ZYNLONTA. Consider prophylactic granulocyte colony-stimulating factor administration as applicable [see Dosage and Administration (2.3)].
5.3 Infections
Fatal and serious infections, including opportunistic infections, occurred in patients treated with ZYNLONTA. Grade 3 or higher infections occurred in 10% of patients, with fatal infections occurring in 2%. The most frequent Grade ≥3 infections included sepsis and pneumonia [see Adverse Reactions (6.1)].
Monitor for any new or worsening signs or symptoms consistent with infection. For Grade 3 or 4 infection, withhold ZYNLONTA until infection has resolved [see Dosage and Administration (2.3)].
5.4 Cutaneous Reactions
Serious cutaneous reactions occurred in patients treated with ZYNLONTA. Grade 3 cutaneous reactions occurred in 4% and included photosensitivity reaction, rash (including exfoliative and maculo-papular), and erythema [see Adverse Reactions (6.1)].
Monitor patients for new or worsening cutaneous reactions, including photosensitivity reactions. Withhold ZYNLONTA for severe (Grade 3) cutaneous reactions until resolution [see Dosage and Administration (2.3)]. Advise patients to minimize or avoid exposure to direct natural or artificial sunlight including exposure through glass windows. Instruct patients to protect skin from exposure to sunlight by wearing sun-protective clothing and/or the use of sunscreen products. If a skin reaction or rash develops, dermatologic consultation should be considered
[see Nonclinical Toxicology (13)].
5.5 Embryo-Fetal Toxicity
Based on its mechanism of action, ZYNLONTA can cause embryo-fetal harm when administered to a pregnant woman because it contains a genotoxic compound (SG3199) and affects actively dividing cells.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ZYNLONTA and for 10 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with ZYNLONTA, and for 7 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
Effusion and Edema [see Warnings and Precautions (5.1)]
Myelosuppression [see Warnings and Precautions (5.2)]
Infections [see Warnings and Precautions (5.3)]
Cutaneous Reactions [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to ZYNLONTA as a single agent at an initial dose of 0.15 mg/kg in 215 patients with DLBCL in studies ADCT-402-201 (LOTIS-2) and ADCT-402-101, which includes 145 patients from LOTIS-2 treated with 0.15 mg/kg x 2 cycles followed by 0.075 mg/kg for subsequent cycles. Among 215 patients who received ZYNLONTA, the median number of cycles was 3 (range 1 to 15) with 58% receiving three or more cycles and 30% receiving five or more cycles.
In this pooled safety population of 215 patients, the most common (>20%) adverse reactions, including laboratory abnormalities, were thrombocytopenia, increased gamma glutamyl transferase, neutropenia, anemia, hyperglycemia, transaminase elevation, fatigue, hypoalbuminemia, rash, edema, nausea, and musculoskeletal pain.
Relapsed or Refractory Diffuse Large B-Cell Lymphoma
LOTIS-2
The safety of ZYNLONTA was evaluated in LOTIS-2 , an open-label, single-arm clinical trial that enrolled 145 patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), including high-grade B-cell lymphoma, after at least two prior systemic therapies [see Clinical Studies (14.1)]. The trial required hepatic transaminases, including gamma glutamyl transferase (GGT), ≤2.5 times upper limit of normal (ULN), total bilirubin ≤1.5 times ULN, and creatinine clearance ≥60 mL/min. Patients received ZYNLONTA 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles and received treatment until progressive disease or unacceptable toxicity. Among the 145 patients, the median number of cycles received was 3, with 34% receiving 5 or more cycles.
The median age was 66 years (range 23 to 94), 59% were male, and 94% had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1. Race was reported in 97% of patients; of these patients, 90% were White, 3% were Black, and 2% were Asian.
Serious adverse reactions occurred in 28% of patients receiving ZYNLONTA. The most common serious adverse reactions that occurred in ≥2% receiving ZYNLONTA were febrile neutropenia, pneumonia, edema, pleural effusion, and sepsis. Fatal adverse reactions occurred in 1%, due to infection.
Permanent treatment discontinuation due to an adverse reaction of ZYNLONTA occurred in 19% of patients. Adverse reactions resulting in permanent discontinuation of ZYNLONTA in ≥2% were gamma-glutamyltransferase increased, edema, and effusion.
Dose reductions due to an adverse reaction of ZYNLONTA occurred in 8% of patients. Adverse reactions resulting in dose reduction of ZYNLONTA in ≥4% was gamma-glutamyltransferase increased.
Dosage interruptions due to an adverse reaction occurred in 49% of patients receiving ZYNLONTA. Adverse reactions leading to interruption of ZYNLONTA in ≥5% were gamma-glutamyltransferase increased, neutropenia, thrombocytopenia, and edema.
Table 1 summarizes the adverse reactions in LOTIS-2.
Table 1: Adverse Reactions (≥10%) in Patients with Relapsed or Refractory DLBCL who received ZYNLONTA in LOTIS-2
Adverse Reaction |
ZYNLONTA (N=145) |
|
|---|---|---|
| All Grades (%) | Grades 3 or 4 (%) | |
| General Disorders and Administration Site Conditions | ||
| Fatigueb | 38 | 1a |
| Edemac | 28 | 3a |
| Skin and Subcutaneous Tissue Disorders | ||
| Rashd | 30 | 2a |
| Pruritus | 12 | 0 |
| Photosensitivity reaction | 10 | 2a |
| Gastrointestinal Disorders | ||
| Nausea | 23 | 0 |
| Diarrhea | 17 | 2a |
| Abdominal paine | 14 | 3 |
| Vomiting | 13 | 0 |
| Constipation | 12 | 0 |
| Musculoskeletal and Connective Tissue Disorders | ||
| Musculoskeletal painf | 23 | 1a |
| Metabolism and Nutrition Disorders | ||
| Decreased appetite | 15 | 0 |
| Respiratory Disorders | ||
| Dyspneag | 13 | 1a |
| Pleural effusion | 10 | 2a |
| Infection | ||
| Upper respiratory tract infectionh | 10 | <1a |
- No Grade 4 adverse reactions occurred
- Fatigue includes fatigue, asthenia, and lethargy
- Edema includes edema, face edema, generalized edema, peripheral edema, ascites, fluid overload, peripheral swelling, swelling, and swelling face
- Rash includes rash, rash erythematous, rash maculopapular, rash pruritic, rash pustular, erythema, generalized erythema, dermatitis, dermatitis acneiform, dermatitis bullous, dermatitis exfoliative generalized, and palmar-plantar erythrodysesthesia syndrome
- Abdominal pain includes abdominal pain, abdominal discomfort, abdominal pain lower, and abdominal pain upper
- Musculoskeletal pain includes musculoskeletal pain, musculoskeletal chest pain, musculoskeletal discomfort, back pain, limb discomfort, myalgia, neck pain, non-cardiac chest pain, and pain in extremity
- Dyspnea includes dyspnea, and dyspnea exertional
- Upper respiratory tract infection includes upper respiratory tract infection, upper respiratory tract congestion, nasopharyngitis, rhinitis, rhinovirus infection, and sinusitis
Clinically relevant adverse reactions in <10% of patients (all grades) who received ZYNLONTA included:
- Blood and lymphatic system disorders: Febrile neutropenia (3%)
- Cardiac disorders: Pericardial effusion (3%)
- Infections: Pneumoniaa (5%), sepsisb (2%)
- Skin and subcutaneous disorders: Hyperpigmentation (4%)
- General disorders: Infusion site extravasation (<1%)
- Pneumonia includes pneumonia and lung infection
- Sepsis includes sepsis, escherichia sepsis, and septic shock
Selected Other Adverse Reactions
- Inflammatory-related conditions were reported in 3% of patients in LOTIS-2, including pericarditis, pneumonitis, pleuritis, and dermatitis.
Table 2 summarizes the laboratory abnormalities in LOTIS-2.
Table 2: Select Laboratory Abnormalities (≥10%) That Worsened from Baseline in Patients with Relapsed or Refractory DLBCL Who Received ZYNLONTA in LOTIS-2
| Laboratory Abnormality | ZYNLONTAa | |
|---|---|---|
| All Grades (%) |
Grade 3 or 4 (%) |
|
| Hematologic | ||
| Platelets decreased | 58 | 17 |
| Neutrophils decreased | 52 | 30 |
| Hemoglobin decreased | 51 | 10b |
| Chemistry | ||
| GGT increased | 57 | 21 |
| Glucose increased | 48 | 8 |
| AST increased | 41 | <1b |
| Albumin decreased | 37 | <1b |
| ALT increased | 34 | 3 |
- The denominator used to calculate the rate varied from 143 to 145 based on the number of patients with a baseline value and at least one post-treatment value.
- No Grade 4 adverse reactions occurred
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of ZYNLONTA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Skin and Subcutaneous Tissue Disorders: telangiectasia, blister, rash vesicular
8 Use in Specific Populations
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, ZYNLONTA can cause embryo-fetal harm when administered to a pregnant woman, because it contains a genotoxic compound (SG3199) and affects actively dividing cells [see Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.1)]. There are no available data on the use of ZYNLONTA in pregnant women to evaluate for drug-associated risk. No animal reproduction studies were conducted with ZYNLONTA. Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Animal reproductive or developmental toxicity studies were not conducted with loncastuximab tesirine-lpyl. The cytotoxic component of ZYNLONTA, SG3199, crosslinks DNA, is genotoxic, and is toxic to rapidly dividing cells, suggesting it has the potential to cause embryotoxicity and teratogenicity.
8.2 Lactation
Risk Summary
There is no data on the presence of loncastuximab tesirine-lpyl or SG3199 in human milk, the effects on the breastfed child, or milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with ZYNLONTA and for 3 months after the last dose.
8.3 Females and Males of Reproductive Potential
ZYNLONTA can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiating ZYNLONTA.
Contraception
Females
Advise women of reproductive potential to use effective contraception during treatment and for 10 months after the last dose.
Males
Because of the potential for genotoxicity, advise males with female partners of reproductive potential to use effective contraception during the treatment with ZYNLONTA and for 7 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Males
Based on the results from animal studies, ZYNLONTA may impair fertility in males. The effects were not reversible in male cynomolgus monkeys during the 12-week drug-free period [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of ZYNLONTA in pediatric patients have not been established.
8.5 Geriatric Use
Of the 145 patients with large B-cell lymphoma who received ZYNLONTA in clinical trials, 55% were 65 years of age and older, while 14% were 75 years of age and older [see Clinical Studies (14.1)]. No overall differences in safety or effectiveness were observed between these patients and younger patients.
8.6 Hepatic Impairment
No dose adjustment is recommended for patients with mild hepatic impairment (total bilirubin ≤ upper limit of normal [ULN] and aspartate aminotransferase (AST) > ULN or total bilirubin > 1 to 1.5 × ULN and any AST). Monitor patients with mild hepatic impairment for potential increased incidence of adverse reactions and modify the ZYNLONTA dosage in the event of adverse reactions [see Dosage and Administration (2.3)].
ZYNLONTA has not been studied in patients with moderate or severe hepatic impairment (total bilirubin > 1.5 × ULN and any AST) [see Clinical Pharmacology (12.3)].
11 Description
Loncastuximab tesirine-lpyl is a CD19-directed antibody and alkylating agent conjugate, consisting of a humanized IgG1 kappa monoclonal antibody conjugated to SG3199, a pyrrolobenzodiazepine (PBD) dimer cytotoxic alkylating agent, through a protease-cleavable valine-alanine linker. SG3199 attached to the linker is designated as SG3249, also known as tesirine.
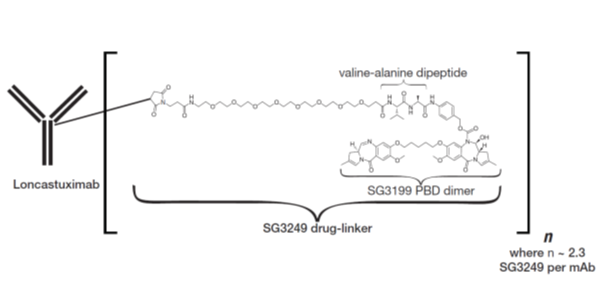
Loncastuximab tesirine-lpyl has an approximate molecular weight of 151 kDa. An average of 2.3 molecules of SG3249 are attached to each antibody molecule. Loncastuximab tesirine-lpyl is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
ZYNLONTA (loncastuximab tesirine-lpyl) for injection is supplied as a sterile, white to off-white, preservative-free, lyophilized powder, which has a cake-like appearance, for intravenous infusion after reconstitution and dilution. Each single-dose vial delivers 10 mg of loncastuximab tesirine-lpyl, L-histidine (2.8 mg), L-histidine monohydrochloride (4.6 mg), polysorbate 20 (0.4 mg), and sucrose (119.8 mg). After reconstitution with 2.2 mL Sterile Water for Injection, USP, the final concentration is 5 mg/mL with a pH of approximately 6.0.
12 Clinical Pharmacology
12.1 Mechanism of Action
Loncastuximab tesirine-lpyl is an antibody-drug conjugate (ADC) targeting CD19. The monoclonal IgG1 kappa antibody component binds to human CD19, a transmembrane protein expressed on the surface of cells of B-lineage origin. The small molecule component is SG3199, a PBD dimer and alkylating agent.
Upon binding to CD19, loncastuximab tesirine-lpyl is internalized followed by release of SG3199 via proteolytic cleavage. The released SG3199 binds to the DNA minor groove and forms highly cytotoxic DNA interstrand crosslinks, subsequently inducing cell death. Loncastuximab tesirine-lpyl had anticancer activity in animal models of lymphoma.
12.2 Pharmacodynamics
Higher loncastuximab tesirine-lpyl exposure in Cycle 1 was associated with higher incidence of some Grade ≥2 adverse reactions, including skin and nail reactions, liver function test abnormalities and increased gamma-glutamyl transferase. Lower loncastuximab tesirine-lpyl exposure in Cycle 1 was associated with lower efficacy over the dose range of 0.015-0.2 mg/kg (0.1 to 1.33 times the maximum recommended dose).
Cardiac Electrophysiology
At the maximum recommended therapeutic dose of 0.15 mg/kg during Cycle 1 and Cycle 2, loncastuximab tesirine-lpyl does not cause large mean increases (i.e., >20 msec) in the QTc interval.
12.3 Pharmacokinetics
The exposure of loncastuximab tesirine-lpyl at the approved recommended dosage in Cycle 2 and at steady state is shown in Table 3. Loncastuximab tesirine-lpyl steady state Cmax was 28.2% lower than the Cmax after the first dose. The time to reach steady state was 105 days.
Table 3: Loncastuximab Tesirine-lpyl Exposure Parametersa
|
Time |
Cmax (ng/mL) |
AUCtau (ng•hr/mL) |
|---|---|---|
Cycle 2 |
2,911 (35.3%) |
21,665 (54.1%) |
Steady state |
1,776 (32.1%) |
16,882 (38.2%) |
Cmax = Maximum observed serum concentration; AUCtau = Area under curve over the dosing interval.
a Data presented as mean and coefficient of variation (CV %)
Distribution
The mean (CV%) of loncastuximab tesirine-lpyl volume of distribution was 7.11 (26.6%) L.
Elimination
The mean (CV%) of loncastuximab tesirine-lpyl clearance decreased with time from 0.499 L/day (89.3%) after a single dose to 0.275 L/day (38.2%) at steady state. The mean (standard deviation) half-life of loncastuximab tesirine-lpyl was 20.8 (7.06) days at steady state.
Metabolism
The monoclonal antibody portion of loncastuximab tesirine-lpyl is expected to be metabolized into small peptides by catabolic pathways. The small molecule cytotoxin, SG3199, is metabolized by CYP3A4/5 in vitro.
Excretion
The major excretion pathways of SG3199 have not been studied in humans. SG3199 is expected to be minimally renally excreted.
Specific Populations
No clinically significant differences in the pharmacokinetics of loncastuximab tesirine-lpyl were observed based on age (20-94 years), sex, race (White vs. Black), body weight (42.1 to 160.5 kg), ECOG status (0 to 2) or mild to moderate renal impairment (CLcr 30 to <90 mL/min using the Cockcroft-Gault equation).
The effect of severe renal impairment (CLcr 15 to 29 mL/min), and end-stage renal disease with or without hemodialysis on loncastuximab tesirine-lpyl pharmacokinetics is unknown.
Patients with Hepatic Impairment
Mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN, or total bilirubin >1 to 1.5 × ULN and any AST) may increase the exposure of unconjugated SG3199, however there was no clinically significant effect on loncastuximab tesirine-lpyl pharmacokinetics. The effect of moderate (total bilirubin >1.5 to ≤3 × ULN and any AST) or severe (total bilirubin >3 ULN and any AST) hepatic impairment on loncastuximab tesirine-lpyl pharmacokinetics is unknown.
Drug Interaction Studies
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: SG3199 does not inhibit CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4/5 at clinically relevant unconjugated SG3199 concentrations.
Transporter Systems: SG3199 is a substrate of P-glycoprotein (P-gp), but not a substrate of breast cancer resistance protein (BCRP), organic anion-transporting polypeptide (OATP)1B1, or organic cation transporter (OCT)1.
SG3199 does not inhibit P-gp, BCRP, OATP1B1, OATP1B3, organic anion transporter (OAT)1, OAT3, OCT2, OCT1, multi-antimicrobial extrusion protein (MATE)1, MATE2-K, or bile salt export pump (BSEP) at clinically relevant unconjugated SG3199 concentrations.
12.6 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies to loncastuximab tesirine-lpyl in other studies or to other products may be misleading.
In LOTIS-2, 0 of 134 patients tested positive for antibodies against loncastuximab tesirine-lpyl after treatment. The potential effect of anti-drug antibodies to ZYNLONTA on pharmacokinetics, efficacy, or safety is unknown.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with loncastuximab tesirine-lpyl or SG3199.
SG3199 was genotoxic in an in vitro micronucleus test and a chromosome aberration assay using human lymphocytes through a clastogenic mechanism. These results are consistent with the pharmacological effect of SG3199 as a covalent DNA crosslinking agent. Results of a bacterial reverse mutation assay (Ames test) were inconclusive due to cytotoxicity.
Fertility studies have not been conducted with loncastuximab tesirine-lpyl. Results from repeat-dose toxicity studies with intravenous administration of loncastuximab tesirine-lpyl in cynomolgus monkeys indicate the potential for impaired male reproductive function and fertility. Administration of loncastuximab tesirine-lpyl to cynomolgus monkeys every 3 weeks at 0.6 mg/kg for a total of 2 doses, or every 3 weeks at 0.3 mg/kg for 13 weeks resulted in adverse findings that included decreased weight and/or size of the testes and epididymis, atrophy of the seminiferous tubules, germ cell degeneration, and/or reduced sperm content. The dose of 0.3 mg/kg in animals results in an exposure (AUC) that is approximately 3 times the exposure at the maximum recommended human recommended dose [MRHD] of 0.15 mg/kg. Findings were not reversible at the end of the 12-week recovery period following 4 or 13 weeks of dosing.
13.2 Animal Toxicology and/or Pharmacology
Inflammatory-mediated toxicities associated with PBDs have been observed at low incidence in animals. In repeat-dose toxicity studies in cynomolgus monkeys, administration of loncastuximab tesirine-lpyl was associated with potential inflammatory-mediated toxicities, including in the lungs and kidneys. Renal toxicity including increased kidney weights and nephropathy with variable inflammation and fibrosis that was reversible was observed in monkeys. Black skin spots potentially related to phototoxicity were observed and were still present after the 12-week treatment-free period.
14 Clinical Studies
14.1 Relapsed or Refractory Diffuse Large B-cell Lymphoma
The efficacy of ZYNLONTA was evaluated in LOTIS-2 (NCT03589469), an open-label, single-arm trial in 145 adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) after at least 2 prior systemic regimens. The trial excluded patients with bulky disease and active central nervous system lymphoma. Patients received ZYNLONTA 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles and received treatment until progressive disease, or unacceptable toxicity.
Of the 145 patients enrolled, the median age was 66 years (range 23 to 94), 59% male, and 94% had an ECOG performance status of 0 to 1. Race was reported in 97% of patients; of these patients, 90% were White, 3% were Black, and 2% were Asian. The diagnosis was DLBCL not otherwise specified (NOS) in 88% (including 20% with DLBCL arising from low-grade lymphoma) and high-grade B-cell lymphoma in 7%. The median number of prior therapies was 3 (range 2 to 7), 63% with refractory disease, 17% with prior stem cell transplant, and 9% with prior chimeric antigen receptor (CAR) T-cell therapy.
Efficacy was established on the basis of overall response rate (ORR) as assessed by an Independent Review Committee (IRC) using Lugano 2014 criteria (Table 4). The median follow-up time was 7.3 months (range 0.3 to 20.2).
Table 4: Efficacy Results in Patients with Relapsed or Refractory DLBCL
|
Efficacy Parameter |
ZYNLONTA |
|---|---|
| Overall response rate by IRCa, (95% CI) | 48.3% (39.9, 56.7) |
| Complete response rate (95% CI) | 24.1% (17.4, 31.9) |
| Partial response rate (95% CI) | 24.1% (17.4, 31.9) |
| Duration of overall responseb | N = 70 |
| Median (95% CI), months | 10.3 (6.9, NE) |
CI = confidence interval, NE = not estimable
aIRC = independent review committee using Lugano 2014 criteria
bOf 70 patients with objective response, 25 (36%) were censored prior to 3 months. Twenty-six percent of responders had a duration of response ≥6 months
The median time to response was 1.3 months (range 1.1 to 8.1).
15 References
- "OSHA Hazardous Drugs." OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html
16 How Supplied/Storage Handling
How Supplied
ZYNLONTA (loncastuximab tesirine-lpyl) for injection is a sterile, preservative-free, white to off-white lyophilized powder, which has a cake-like appearance, supplied in a single-dose vial for reconstitution and further dilution. Each carton (NDC 79952-110-01) contains one 10 mg single-dose vial.
Storage and Handling
Store refrigerated at 2°C to 8°C (36°F to 46°F) in original carton to protect from light. Do not use beyond the expiration date shown on the carton. Do not freeze. Do not shake.
Special Handling
ZYNLONTA is a hazardous drug. Follow applicable special handling and disposal procedures.1
Any unused drug product or waste material should be disposed in accordance with local requirements.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
- Effusion and Edema: Advise patients to contact their healthcare provider if they experience swelling, weight gain, shortness of breath, or difficult, labored breathing [see Warnings and Precautions (5.1)].
- Myelosuppression: Advise patients to immediately contact their healthcare provider for a fever of 100.4°F (38°C) or greater, or signs or symptoms of bruising or bleeding. Advise patients of the need for periodic monitoring of blood counts [see Warnings and Precautions (5.2)].
- Infections: Advise patients to contact their healthcare provider for signs or symptoms of infection, including fever, chills, weakness and/or difficulty breathing [see Warnings and Precautions (5.3)].
- Cutaneous Reactions: Advise patients that skin reaction or rash can occur. Patients should be directed to minimize or avoid exposure to direct natural or artificial sunlight, including sunlight exposure through glass windows. Patients should be instructed to protect skin from exposure to sunlight by wearing sun-protective clothing and/or the use of sunscreen products [see Warnings and Precautions (5.4)].
- Embryo-Fetal Toxicity:
- Advise pregnant women of the potential risk to a fetus. Advise female patients of reproductive potential to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with ZYNLONTA [see Use in Specific Populations (8.1)].
- Advise women of reproductive potential to use effective contraception during treatment with ZYNLONTA and for 10 months after the last dose.
- Advise male patients with female partners of reproductive potential, to use effective contraception during treatment with ZYNLONTA and for 7 months after the last dose [see Warnings and Precautions (5.5) and Use in Specific Populations (8.1, 8.3)].
- Lactation: Advise women not to breastfeed during treatment with ZYNLONTA and for 3 months after the last dose [see Use in Specific Populations (8.2)].
Manufactured by:
ADC Therapeutics SA
Route de la Corniche 3B
1066 Epalinges, Switzerland
U.S. license No. 2166
Distributed by:
ADC Therapeutics America
Murray Hill,
New Jersey 07974
For more information, go to www.ZYNLONTA.com or call 1-855-690-0340
ZYNLONTA is a registered trademark of ADC Therapeutics SA
PAB is a registered trademark of B. Braun Medical Inc.
Patient Information
ZYNLONTA® (zin lon’ tah)
(loncastuximab tesirine-lpyl)
for injection, for intravenous use
What is ZYNLONTA?
ZYNLONTA is a prescription medicine used to treat adults with certain types of large B-cell lymphoma that has come back (relapsed) or that did not respond to previous treatment (refractory), who have already received two or more treatments for their cancer.
Before you receive ZYNLONTA, tell your healthcare provider about all of your medical conditions, including if you:
- have an active infection or have had one recently.
- have liver problems.
- are pregnant or plan to become pregnant. ZYNLONTA can harm your unborn baby.
Females who can become pregnant:- Your healthcare provider may do a pregnancy test before starting treatment with ZYNLONTA.
- You should use effective birth control (contraception) during treatment with ZYNLONTA and for 10 months after the last dose of ZYNLONTA. Talk to your healthcare provider about effective birth control. Tell your healthcare provider right away if you become pregnant or think that you are pregnant during treatment with ZYNLONTA.
- You should use effective birth control (contraception) during treatment with ZYNLONTA and for 7 months after the last dose of ZYNLONTA.
- are breastfeeding or plan to breastfeed. It is not known if ZYNLONTA passes into breast milk. Do not breastfeed during treatment with ZYNLONTA and for 3 months after the last dose of ZYNLONTA.
How will I receive ZYNLONTA?
- ZYNLONTA is given to you by your healthcare provider as an intravenous (IV) infusion into your vein over 30 minutes.
- ZYNLONTA is usually given every 3 weeks.
- Your healthcare provider may give you medicine before each infusion to decrease your chance of side effects.
- Your healthcare provider may stop your treatment, delay your treatment, or change your dose of ZYNLONTA if you have severe side effects.
- Your healthcare provider should do blood tests regularly to check for side effects of ZYNLONTA.
- Your healthcare provider will decide how many treatments you need.
What should I avoid while receiving ZYNLONTA?
Avoid or limit your exposure to sunlight, including sunlight through glass, such as buildings or vehicle windows and artificial sunlight such as sunlamps or tanning beds. Exposure to sunlight during treatment with ZYNLONTA can cause skin reaction or rash. Use sun protection measures such as sunscreen and wear loose-fitting clothes that cover your skin while out in sunlight.What are the possible side effects of ZYNLONTA?
ZYNLONTA may cause serious side effects, including:
- Fluid retention. Your body may hold too much fluid during treatment with ZYNLONTA. This can be serious. Tell your healthcare provider if you develop new or worsening swelling or puffiness, weight gain, chest pain, shortness of breath, or trouble breathing.
- Low blood cell counts (platelets, red blood cells, and white blood cells). Low blood cell counts are common with ZYNLONTA but can also be serious or severe. Your healthcare provider will monitor your blood counts during treatment with ZYNLONTA. Tell your healthcare provider right away if you get a fever of 100.4°F (38°C) or above, or any bruising or bleeding.
- Infections. Serious infections, including infections that can cause death, have happened in people treated with ZYNLONTA. Tell your healthcare provider right away if you have new or worsening signs or symptoms of infection, including:
- fever
- chills
- flu-like symptoms (cough, tiredness or weakness, and body aches)
- headache
- breathing problems
- cuts or scrapes that are red, warm, swollen or painful
- Skin Reactions. Serious skin reactions have happened in people treated with ZYNLONTA. Tell your healthcare provider if you get new or worsening skin reactions, including sensitivity to sunlight, skin rash, peeling, redness or irritation. You may burn more easily or get severe sunburns. See “What should I avoid while receiving ZYNLONTA?”
The most common side effects of ZYNLONTA include:
|
|
ZYNLONTA may cause fertility problems in males which may affect your ability to father children. Talk to your healthcare provider if this is a concern for you.
These are not all of the possible side effects of ZYNLONTA. Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of ZYNLONTA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your healthcare provider or pharmacist for information about ZYNLONTA that is written for healthcare professionals.
What are the ingredients in ZYNLONTA?
Active ingredient: loncastuximab tesirine-lpyl
Inactive ingredients: L-histidine, L-histidine monohydrochloride, polysorbate 20, and sucrose
Manufactured by: ADC Therapeutics SA, Route de la Corniche 3B, 1066 Epalinges, Switzerland
U.S. license number 2166
Distributed by: ADC Therapeutics America, Murray Hill, New Jersey 07974
ZYNLONTA is a registered trademark of ADC Therapeutics SA
For more information, go to www.ZYNLONTA.com or call 1-855-690-0340
This Patient Information has been approved by the U.S. Food and Drug Administration.
Issued: Oct 2022